Свежие публикации

В этой статье мы разберемся и рассмотрим способы, как узнать было ли сообщение в Instagram прочитано
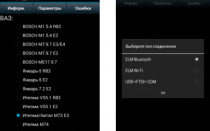
Прошивка телевизора Samsung через USB. Обновление ПО Smart TV с флешки

📱 3G модем для ГУ на "Андроид" Outlander 2012-2015: эксплуатация и дополнительное оборудование

Иногда нам кажется, что в WhatsApp необходимо иметь пароль, но, к сожалению, в приложении WhatsApp нет функции по умолчанию для...

Бывают случаи, когда вы смотрите что-то на своем телефоне и хотите поделиться этим с комнатой или просто увидеть это на...
![Как обновить Инстаграм [+Автообновления]](https://zdrons.ru/wp-content/cache/thumb/92/f4622bd9676ce92_320x200.jpg)
В этой статье мы расскажем вам, как обновить приложение Instagram на устройстве iOS или Android.

📱 Когда при работе в сети Интернет выдаётся "ошибка SSL", то это действительно может обозначать, что кто-то случайно или умышленно...

📱 Что делать, если на телефоне Xiaomi или Redmi появилась реклама прямо на экране блокировки? Инструкция, как ее полностью отключить...

📱 Лучшие онлайн кинотеатры для Андроид ТВ и медиа-приставок. Разборка функционала, возможностей и особенностей. Плюсы и минусы каталогизаторов.

Привет, Хабр! представляю вашему вниманию перевод статьи «Top Programming Languages for Android App Development» автора Mohit Maheshwari. Бурное развитие информационных...

Если у вас есть основания полагать, что ваш смартфон или стационарный телефон может прослушиваться, есть несколько признаков, которые могут подтвердить...

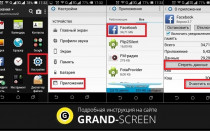
Как установить на Смарт ТВ приложения — какие виды бывают, в чем их особенности и отличия. Как найти нужный виджет...
Показать ещё
Популярные публикации








detector